Quality-by-Design (QbD)
Historical background of Quality-by-Design (QbD)
Traditional product development and manufacturing methods are based on a philosophy of Quality-by-Testing (QbT). In QbT, the quality is assessed after the product has been designed and manufactured. If problems are detected using the QbT approach, it is usually too late to fix the current batch. It is possible that this process must be repeated multiple times before adequate quality is reached.
The modern approach, Quality-by-Design (QbD), introduces quality assessment as early as possible in the product lifecycle (design phase). It was firstly introduced in the guidance “cGMPs for the 21st century” published by the US Food and Drug Administration (FDA) and later in the ICH guidelines Q8-Q12. QbD is a risk-based development approach that introduces the concepts of quality, efficacy and safety over the complete product lifecycle.
Why using QbD?
If used properly, a QbD approach can help address issues early in the product development process and prevent failures at an advanced state of development. Nowadays, projects in pharmaceutical and medical device industries can no longer be carried out without taking into account the recent good practices of development recommended in ICH directives Q8-Q12. Indeed, health authorities require applicants to better understand the complex relationships that exist between product efficiency, quality assurance, toxicity risks and regulatory compliance.
The overarching goal of QbD is to improve biological therapeutics’ safety and efficacy, while additional benefits include increasing the efficiency of the manufacturing process, which reduces cost, and the potential for faster regulatory approval of new drugs. QbD has been successfully used in different areas including pharmaceutical industry. The TBMED project aims at extending its application to the safe development of medical devices.
Main steps of QbD
QbD is a systematic development approach that begins with predefined objectives and emphasizes product and process understanding, based on sound science and quality risk management1. The QbD implementation relies on a large collection of statistical methods and tools from the design of experiments up to the multivariate regression analysis for risk prediction. One key step is to determine the relationship between critical quality/safety attributes (CQA) and some controllable factors associated with the formulation and manufacturing phases (Critical Material Attributes (CMA) and Critical Process Parameters (CPP)) in order to identify a parametric region (Design Space) in which the probabilities of compliance with quality and safety specifications are acceptable. The whole QbD cycle integrates several key steps described in Figure 1.
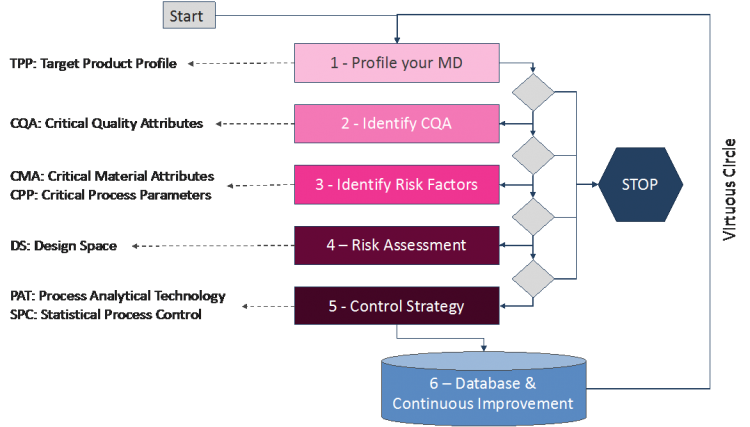
Step 1: Quality Target Product Profiles (QTPP)
The Quality Target Product Profile (QTPP) is a key strategic document that provides a summary of the product under development; the product’s desired characteristics and features; the studies and activities that must be completed to demonstrate the product efficacy, safety and quality; as well as the features of a product that provide a competitive advantage. Each section of the QTPP provides the opportunity to describe the key differentiating features and competitive positioning of the product to be developed.
Step 2: Critical Quality Attributes (CQA)
A Critical Quality Attribute (CQA) is a physical, chemical, biological or microbiological property or characteristics of a nanomaterial that should be within an appropriate limit, range or distribution (specification) to ensure the desired product quality, efficacy or safety. A CQA can be a size distribution, targeting efficacy, encapsulation efficacy, polydispersity index, amount of release, etc. It is recommended to measure CQAs with standards of protocol to better control experimental errors during experimentations. CQAs cannot be controlled directly but indirectly through input factors divided into two categories: CMAs and CPPs (see Step 3).
Step 3: Critical Material Attributes (CMA) and Critical Process Parameters (CPP)
“Material” means raw material, starting materials, reagents, solvents, process aids, intermediates, etc. while “Attribute” means a physical, chemical, biological or microbiological property or characteristics of this material that can be measured. It could be the particle morphology, ingredient concentration and type, ingredient ratio, drug loading, surfactant concentration, aqueous solubility, pH etc. A Material Attribute is critical when it is very likely to impact a CQA.
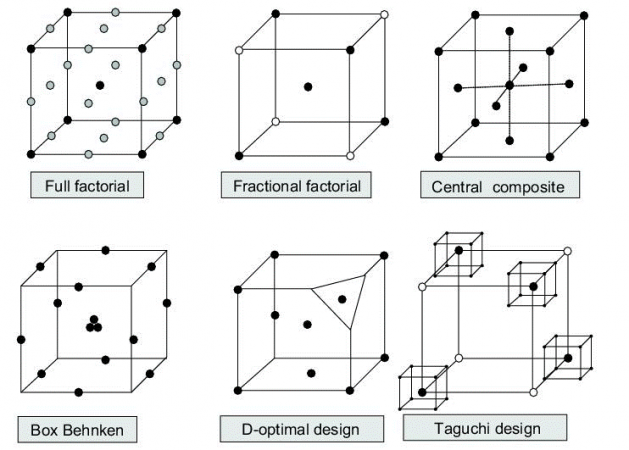
A Critical Process Parameter (CPP) is another category of risk factors associated with manufacturing variables. As CMAs, CPPs can be directly controlled by experimenters and have a significant impact on CQAs. Their identification is an important step of QbD to determine which parameters shall be used as input variables to control CQAs and maintain them in the specified range for quality and safety considerations. As illustrated in Figure 2, the main steps of QbD require the application of statistical methods for the design of experiments. Those techniques allow to get the most informative data by minimizing the number of trials and finally to determine cause-effect mathematical relationships between CMAs CPPs (causes) and CQAs (consequences).
Step 4: Design Space
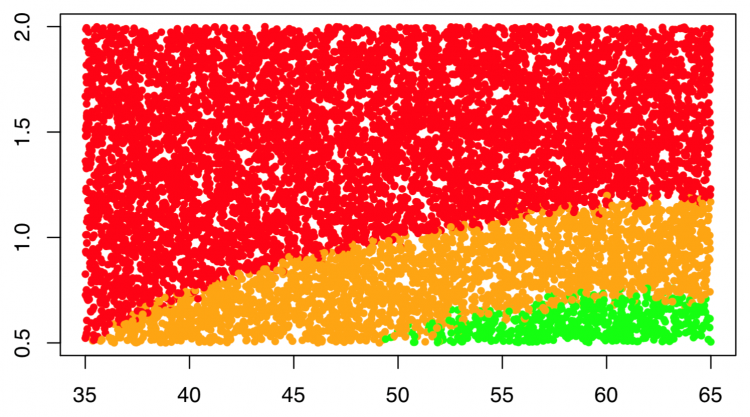
The Design Space is a key concept in Quality by Design, which is deduced from a Response-Surface model identified from experimental datasets. It allows manufacturers to explore combinations of multiple variables (CMAs and CPPs) using appropriate statistical tools. The Design Space provides a top-down and multivariate view of the process, enabling operators and/or control systems to make the right changes to maintain optimal quality. Figure 3 shows an example of a Design Space where three main regions are defined:
- Normal Operating Region (NOR): this is the desired state in which the highest quality product exists. In this case the NOR is represented in green and corresponds to the region of critical parameters for which the probability to fulfill the CQA specifications is greater than a threshold chosen by the developers
- Proven Acceptance Region (PAR): the risk not to comply with the CQA specifications is still acceptable but process adjustments should be made to return operation to NOR levels.
- Out Of Specifications (OOS): the state of the product is not acceptable. Investigations must take place to determine the reasons and decide on a course of action.
Step 5: Process Analytical Technology (PAT) & Control Strategy
To implement QbD effectively, we must be able to make timely measurements that inform about the location (state) of the current product in the Design Space. PAT means “Process Analytical Technology” and consists in technologies able to measure, on-line or off-line, some CMA or CPP variables during processing. When a PAT system detects a process drift or change within the process’ design space, actions can be taken so that the desired state is always maintained. Those actions depend on a control strategy such as statistical process control.
Step 6: Data Management (DM) & Continuous Improvement
During manufacturing, off-line/on-line measurement and testing are carried out and production data are aggregated into a database. Data management includes data identification, acquisition, synchronization, modeling, coordination, organization, distribution, architecting and storage. Those data are finally iteratively used in the QbD cycle to refine the knowledge, improve the accuracy of predictions and therefore the quality of products. Accordingly, it is a key component to implement continuous improvement. Without careful data management systems, handling and analysis of data can be very painful. Data integrity is also an essential element for creating trustworthy records that form the basis for knowledge management. Indeed, breaches in data integrity can have negative consequences and may lead to patient injury or even death.
If you are interested to learn more about the QbD approach, get in contact with TBEMD partner CYBERnano.
References
- T. Bastogne. Quality-by-design of Nanopharmaceuticals – A state of the art. Nanomedicine: Nanotechnology, Biology and Medicine, 13(7):2151–2157, 2017.
- B. Halamoda-Kenzaoui, S. Baconnier, T. Bastogne, D. Bazile, P. Boisseau, G. Borchard, S. E. Borgos, L. Calzolai, K. Cederbrant, G. di Felice, T. Di Francesco, M. Dobrovolskaia, R. Gaspar, B. Gracia, V. A. Hackley, L. Leyens, N. Liptrott, M. Park, A. Patri, G. Roebben, M. Roesslein, R. Thürmer, P. U. López, V. Zuang, and S. Bremer-Hoffmann. Bridging communities in the field of nanomedicine. Regulatory Toxicology and Pharmacology, 2019.
ICH Q8R2 Pharmaceutical Development. Geneva, Switzerland, International conference on harmonisation of technical require- ments for registration of pharmaceuticals for human use. 2009. http://www.ich.org/LOB/media/MEDIA4986.pdf. ↩